

Molti fabbricanti di nuovi dispositivi medici, in vista della scadenza del 26 maggio 2021 che prevede la fine del periodo transitorio in cui è ancora possibile marcare CE con la vecchia direttiva MDD , si stanno affrettando per rendere conformi i loro nuovi prodotti a tale direttiva i cui certificati perdono di validità al più tardi il il 27 maggio 2024
Tuttavia l’articolo 120 del Regolamento 2017/745 “Disposizioni Transitorie” pone alcune condizioni affinché la marcatura MDD dei dispositivi continui a valere e precisamente :
- che si applichino le prescrizioni del Regolamento MDR in materia di sorveglianza post-commercializzazione, sorveglianza del mercato, vigilanza, registrazione degli Operatori economici e dei dispositivi sostituendo le corrispondenti prescrizioni relative alla vecchia direttiva MDD
- Che non vi siano significativi cambiamenti nella progettazione e nella destinazione d’uso
Ora, questa seconda condizione, mentre per i fabbricanti di manufatti può essere un vincolo nella maggior parte dei casi più teorico che pratico in quanto difficilmente un prodotto cambia in un breve lasso di tempo le proprie modalità operative e soprattutto la propria destinazione d’uso, diverso è il discorso per le Applicazioni Software utilizzabili a vario titolo come Dispositivi Medici
Infatti coloro che hanno a che fare con prodotti software, in qualunque settore siano utilizzati, sanno che tali Applicazioni sono sempre un cantiere aperto e che modifiche di varia natura sono inserite in modo continuo e sistematico e che possono avere influenza sull’operatività e a volte anche sulla tipologia di utilizzo soprattutto allorquando si inseriscono nuove funzionalità
La realizzazione di modifiche all’interno di un’applicazione software può avere due grosse conseguenze:
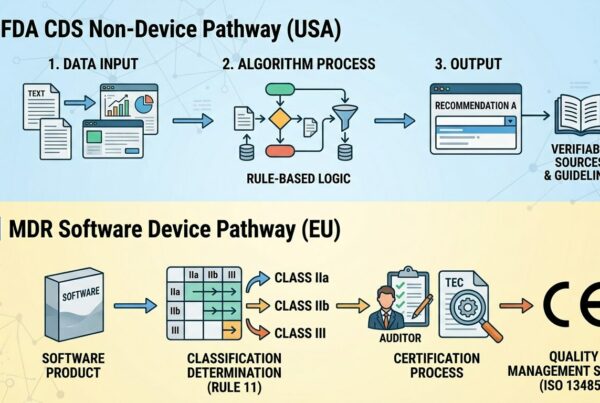
- portare l’applicazione software (se Stand Alone), ma anche lo stesso Dispositivo Elettromedicale ospite (se firmware/embedded), verso una classe di rischio più severa (ad esempio da una classe IIa ad una IIb)
- pur non inducendo un cambio di classe di rischio può comunque rientrare nei vincoli del art. 120 che fanno progettualmente decadere la Marcatura MDD con la necessità di ricertificare il prodotto come MDR
Per questo secondo caso è necessario individuare quali tipologie di modifiche software rientrino nelle condizioni definite all’art. 120 come “cambiamenti significativi nella progettazione” e nella “destinazione d’uso”
Il documento MDCG 2020-3 riporta alcuni criteri che possono aiutare i progettisti a valutare le possibili conseguenze di modifiche progettuali. Tale documento infatti dà alcune indicazioni (non esaustive) di quali tipologie di interventi definiti Maggiori possano essere ritenute significative, rispetto a quanto richiesto dall’ MDR, e quali possano essere ritenute Minori non determinando un cambio di stato dal punto di vista regolatorio
E’ da tener presente che l’eventuale mancato adeguamento di Marcatura di Applicazioni Software come dispositivi Medici potrebbe portare a situazioni sanzionatorie non solo per i fabbricanti, per aver immesso sul mercato prodotti non marcati adeguatamente, ma anche per gli utilizzatori qualora sia dimostrata la loro consapevolezza del fatto che le modifiche apportate al prodotto software acquistato avrebbero dovuto prevedere una ricertificazione.
E’ dunque estremamente importante che, al fine di evitare pericolosi “sconfinamenti”, i processi collegati alle Change Request siano gestiti attraverso procedure scritte del Sistema Qualità che mettano soprattutto in grande evidenza criteri affidabili di valutazione delle modifiche che si vogliono apportare (collegate ad esempio ad una robusta Risk Analysis) in modo che i progettisti software siano consapevoli delle eventuali conseguenze regolatorie a cui stanno andando incontro

Commenti recenti