
Uno dei settori informatici che sta avendo in questo periodo un grande sviluppo è quello relativo alle Applicazioni Software da impiegare in ambito medico: pensiamo solo alle numerose App utili a monitorare ed elaborare i parametri fisiologici, l’attività fisica, le calorie bruciate, il battito cardiaco, etc.
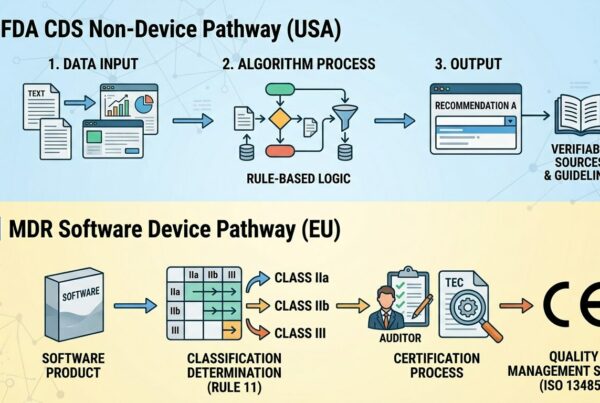
In tale contesto però non appare un compito facile determinare quali, fra tutti gli sviluppi software proposti, possono essere classificati come Dispositivi Medici e quindi rientrare nell’ambito regolatorio che permette loro di essere qualificati come affidabili in termini di rispetto dei requisiti di sicurezza, efficacia e qualità, sulla base delle specifiche classi di rischio
Mentre non si possono nutrire dubbi per quelle Applicazioni Software di tipo firmware o embedded e che risiedono all’interno delle apparecchiature medicali, oppure che, pur risiedendo su dispositivi esterni, sono strettamente collegati ed integrati alle funzionalità hardware e software del Dispositivo, diverso è il discorso per i prodotti software cosiddetti Stand Alone (es. le App)
Per tali tipologie di applicazioni è importante quindi capire quando sono classificabili come Dispositivi Medici e quindi rientrare, ad esempio, nella regolamentazione MDR necessaria alla loro commercializzazione in ambito europeo.
1. CRITERI PER VALUTARE UNA APPLICAZIONE SOFTWARE
SaMD (Software as Medical Device): è definito come un software destinato a essere utilizzato per uno o più scopi medici e che svolgono tali scopi senza essere incorporato in un dispositivo medico al momento della sua immissione sul mercato o della sua messa a disposizione
Non tutte le applicazioni software utilizzate in ambito medicale sono classificabili come SaMD
La linea guida MEDDEV 2.1/6 (Medical Devices: Guidance document. Qualification and Classification of stand-alone software) stabilisce criteri precisi per la valutazione dei software “stand alone” come SaMD e precisamente:
- sia un programma informatico (e non un semplice documento digitale come potrebbe esserlo ad esempio un foglio excel);
- svolga una funzione diversa e ulteriore rispetto alla mera conservazione o trasmissione o ricerca di dati;
- svolga una delle funzioni incluse nella definizione di dispositivo medico (vale a dire diagnosi, prevenzione, controllo o terapia di una malattia o di una ferita o di un handicap; studio, sostituzione o modifica dell’anatomia o di un processo fisiologico; intervento sul concepimento ecc.);
- operi a beneficio di specifici pazienti (ad esempio non dia responsi generali sulla base di valutazioni statistiche)
Alcuni esempi concreti si possono ricavare nel Manual on borderline and classification in the Community Regulatory Framework for medical devices redatto dal gruppo di esperti della Commissione europea e costantemente aggiornato
2. INDIVIDUAZIONE DELLA CLASSE DI APPARTENENZA
Per quelle applicazioni software, che sono state classificate come SaMD e per le quali é necessario ottenere la conformità con la Medical Device Regulation MDR 2017/745, è indispensabile stabilire a quale classe appartengono tali applicazioni.
Il documento di riferimento è, come per qualunque tipologia di Dispositivi Medici, l’allegato VIII del regolamento MDR il quale chiarisce che i software classificati come dispositivi medici rientrano nella classe I ad eccezione di quelli destinati a monitorare i processi fisiologici che rientrano nella classe IIa oppure IIb allorquando oggetto del monitoraggio sono parametri vitali la cui variazione può creare un pericolo immediato per il paziente.
L’iter per l’ottenimento del marchio CE dipende dalla classe di appartenenza ed è quello indicato negli specifici paragrafi ed allegati del regolamento.
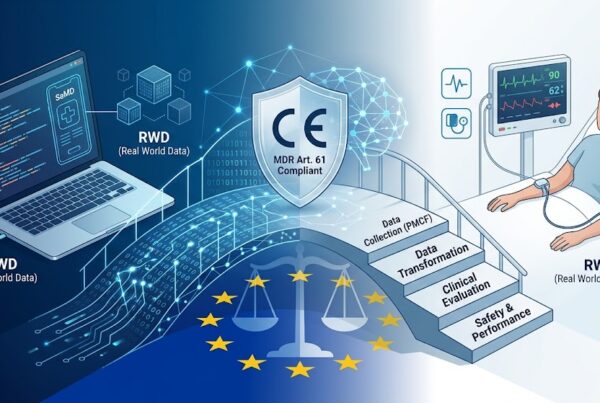
3. LA DOCUMENTAZIONE DI CONFORMITA’ ALL’ MDR
Nel caso di un SaMD il percorso che porta alla conformità del sistema riguarda la realizzazione delle seguenti tipologie di documenti:
A) Documentazione Tecnica che comprende:
- descrizione e Specifiche del Dispositivo, inclusi requisiti di Sicurezza e prestazioni ( Specifiche dei requisiti Software)
- informazioni che devono essere fornite dal fabbricante (Manuale d’Uso e di Installazione)
- Informazioni di Progettazione e Fabbricazione (es. Specifiche funzionali, di disegno, di Prove controlli e collaudi ecc.) inclusa analisi dei rischi e dei benefici e gestione del rischio
- verifiche e convalida del prodotto
B) Documentazione della Sorveglianza post commercializzazione che comprende:
- Piano di sorveglianza ( Gestione dei reclami, Non conformità Gravi o non Gravi, valutazione periodica applicazioni analoghi)
- Processi / procedure di azioni correttive, preventive e di gestione delle modifiche e di aggiornamento delle release)
- Processi / procedure per garantire la identificazione, rintracciabilità e le azioni di sostituzione
- Rapporto Periodico di aggiornamento sulla Sicurezza
C) Dichiarazione di Conformità UE
4. IDENTIFICAZIONE DELL’APPLICAZIONE SOFTWARE
Come ogni Dispositivo Medico anche un SaMD è soggetto ad una metodologia di identificazione che è comune a livello europeo e che ha lo scopo di garantire la massima rintracciabilità nella catena di fornitura facilitando, quando necessario, le opportune operazioni di richiamo di quei dispositivi che dovessero presentare rischi per la sicurezza
Tale identificazione è detta UDI ed è formata da due elementi:
- UDI-DI che è un codice numerico o alfanumerico unico, specifico di un modello di dispositivo ed è anche usato come “chiave di accesso” alle informazioni memorizzate in una banca dati UDI.
- UDI-PI che è un codice numerico o alfanumerico che identifica la specifica unità di produzione del dispositivo (es. il numero di serie, il numero del lotto/della partita, la fabbricazione ecc.)
Nel caso delle applicazioni SaMD la loro identificazione è soggetta alle seguenti regole:
1. Ad ogni nuova versione del SaMD deve essere associato un nuovo identificativo UDI.
2.Ogni volta che vi sia stata una modifica significativa (revisione di tipo maggiore) siamo in presenza di un prodotto nuovo e quindi deve essere identificato con un nuovo UDI-DI. Per modifiche significative si intendono quelle che riguardano:
-
- le prestazioni e l’efficacia originali
- la sicurezza per il paziente e l’utilizzatore oppure l’uso previsto del SaMD
Queste modifiche possono includere algoritmi nuovi o modificati, strutture di database, la piattaforma operativa, l’architettura, nuove interfacce utente o nuovi canali per l’interoperabilità ecc.
3. Ogni volta invece che viene effettuata una modifica minore la nuova revisione dovrebbe essere identificata attraverso un nuovo UDI-PI.
Queste modifiche possono riguardare correzioni di errori, miglioramenti dell’usabilità non collegati però alla sicurezza fisica, interventi sulla sicurezza delle informazioni, miglioramenti dell’efficienza ecc.
5. VALIDAZIONE PER APPLICAZIONI NON SaMD
Alle Applicazioni software attinenti all’ambito della salute e benessere ma non classificabili come dispositivi medici sono invece dedicate le “EU Guidelines on assessment of the reliability of mobile health applications”, anch’esse predisposte dalla Commissione europea.
Si tratta di una valutazione dell’Applicazione software sulla base di una dettagliata Analisi dei Rischi e di una serie di criteri quali :
- Affidabilità : che serve a valutare se l’applicazione è in grado di funzionare correttamente anche sotto diverse circostanze e situazioni
- Stabilità : che serve a valutare come l’applicazione reagisca, durante l’uso, ad esempio alla perdita di rete, perdita di energia ecc.
- Efficacia : che cerca di identificare prove dell’efficacia dell’applicazione nel raggiungimento degli obiettivi dichiarati.
- Usabilità e Accessibilità : che serve a valutare se l’applicazione è utilizzabile dalle persone a cui è indirizzata
- Trasparenza : che indaga sugli elementi che ne hanno permesso la realizzazione (chi lo ha finanziato, per quale motivo, chi detiene i dati dell’utente ecc.)
- Credibilità : che serve a valutare se le caratteristiche funzionali e la metodologia offerta dall’applicazione si basa su appropriata documentazione ed è stata convalidata da organismi autorizzati
- Sicurezza (safety) : che serve a valutare se l’applicazione è impostata in modo appropriato rispetto alle aspettative dell’utente per un funzionamento sicuro
- Sicurezza (Privacy & Security) : che serve a valutare se l’applicazione è stata programmata sulla base di quanto previsto da regolamenti Europei sui dati personali (es. GDPR)
- Desiderabilità : che serve a valutare se l’applicazione come è presentata non porti gli utilizzatori a stancarsi rapidamente dell’applicazione stessa

Commenti recenti